La fonction minimum [5]
On considère une structure comportant N atomes
en positions inconnues
![]() dans la maille, pour laquelle la mesure des intensités de diffraction
X d'un ensemble de réflexions {H} a fournit après
normalisation les modules des facteurs de structure
dans la maille, pour laquelle la mesure des intensités de diffraction
X d'un ensemble de réflexions {H} a fournit après
normalisation les modules des facteurs de structure
![]() .
.
Si on suppose que les atomes ont tous à peu près le même poids, les facteurs de structure normalisés s'écrivent sous la forme simplifiée :
![]()
On note
![]() les triplets de phases invariants dans un changement d'origine, et
les triplets de phases invariants dans un changement d'origine, et
![]() les quadruplets de phases invariants :
les quadruplets de phases invariants :
![]()
![]()
D'un point de vue statistique, si on considère
les vecteurs position
![]() comme
des variables aléatoires indépendantes et uniformément
distribuées, les invariants seront aussi des variables aléatoires.
Pour un couple de réflexions H, K donné, on
peut alors montrer que le cosinus du triplet de phase correspondant
comme
des variables aléatoires indépendantes et uniformément
distribuées, les invariants seront aussi des variables aléatoires.
Pour un couple de réflexions H, K donné, on
peut alors montrer que le cosinus du triplet de phase correspondant
![]() peut être estimé par :
peut être estimé par :
![]() où
où
![]() et I0, I1 sont les fonctions de Bessel modifiées
d'ordre 0 et 1 respectivement ; cette estimation sera d'autant plus valide
que
et I0, I1 sont les fonctions de Bessel modifiées
d'ordre 0 et 1 respectivement ; cette estimation sera d'autant plus valide
que
![]() sera grand.
sera grand.
De manière identique, pour des réflexions
L,
M
et N données, le cosinus du quadruplet
![]() peut être estimé par :
peut être estimé par :
![]() avec
avec
![]() .
.
D'où le choix de la fonction minimum utilisée par SnB :
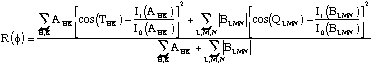
La double somme porte sur les vecteurs du réseau
réciproque H, K associés aux triplets
![]() correspondants aux plus grandes valeurs de
correspondants aux plus grandes valeurs de
![]() et la triple somme sur les vecteurs L, M, N associés
aux quadruplets
et la triple somme sur les vecteurs L, M, N associés
aux quadruplets
![]() négatifs.
Dans ces sommes,
négatifs.
Dans ces sommes,
![]() représente
le poids du triplet
représente
le poids du triplet
![]() et
et ![]() le poids du quadruplet
le poids du quadruplet
![]() .
.
D'après son expression, on constate que R(f) est une mesure de la différence quadratique moyenne entre les cosinus des invariants calculés et leurs valeurs attendues. Le " principe minimum " énoncé par H.Hauptman stipule que :
- pour un nombre suffisant de phases, contraintes à prendre des valeurs consistantes avec une structure atomique, R(f) présente un minimum global lorsque toutes ces phases sont égales à leur vraie valeur.
- lorsque les phases sont égales à leur vraie valeur, R(f) = RT < 1/2, indépendemment du choix de l'origine et de l'énantiomorphe.
- au contraire pour des phases aléatoires on a R(f) = RR > 1/2.
Le fonctionnement de SnB découle directement du principe minimum : il consiste à faire varier les phases dans l'espace réciproque en vue de minimiser la valeur de la fonction minimum, puis à revenir dans l'espace direct pour contraindre les phases à prendre des valeurs consistantes avec une structure atomique. Un nombre (proportionnel à la taille de la structure) de tels cycles est effectué pour tenter de converger vers la solution.
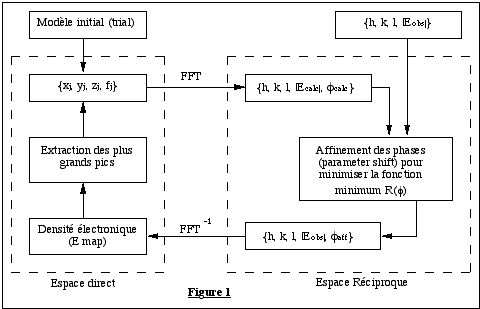
L'algorithme correspondant, schématisé sur la figure 1, comporte donc les étapes suivantes :
- Un modèle initial composé d'un nombre d'atomes inférieur ou égal au nombre recherché est généré ; ces atomes sont placés aléatoirement dans l'unité asymétrique avec la contrainte qu'ils doivent être distants d'au moins 1.2 Å, et ne pas être liés à plus de quatre voisins.
- Un calcul des facteurs de structure normalisés
est effectué pour le modèle courant, et les phases ainsi
obtenues seront affectées aux
![]() observés.
observés.
- L'affinement des phases peut être réalisé
par diverses méthodes, dont la traditionelle formule de la tangente
[11],
mais il est recommandé d'utiliser celle par défaut, dite
" parameter shift ". Les phases sont considérées par ordre
décroissant des valeurs de
![]() associées ; lors de la modification de la ième
phase, la fonction minimum est évaluée pour sa valeur actuelle
fi,
pour
fi
+ Df et pourfi
- Df, où Df
est un incrément fixé (Figure 2). Si R(f)
est minimum pour la valeur actuelle de fi
celle-ci est conservée et on passe à la phase suivante fi+1
. Sinon on choisit le sens de variation
donnant R(f) minimum
(+Df sur la figure
2) ; la valeur retenue de la phase subit de nouveaux déplacements
dans le même sens tant que R(f)
diminue et qu'un nombre maximum de déplacements n'a pas été
dépassé (3 dans l'exemple).
associées ; lors de la modification de la ième
phase, la fonction minimum est évaluée pour sa valeur actuelle
fi,
pour
fi
+ Df et pourfi
- Df, où Df
est un incrément fixé (Figure 2). Si R(f)
est minimum pour la valeur actuelle de fi
celle-ci est conservée et on passe à la phase suivante fi+1
. Sinon on choisit le sens de variation
donnant R(f) minimum
(+Df sur la figure
2) ; la valeur retenue de la phase subit de nouveaux déplacements
dans le même sens tant que R(f)
diminue et qu'un nombre maximum de déplacements n'a pas été
dépassé (3 dans l'exemple).
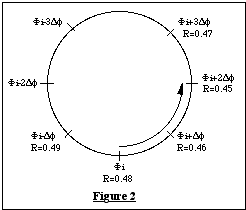
Par exemple sur la figure 2, la nouvelle valeur de fi sera fi + 2 Df. On notera que cette nouvelle valeur remplace immédiatement l'ancienne en mémoire, et est donc utilisée lors de l'affinement des phases suivantes. Lorsque le programme a parcouru toutes les phases, il recommence ce processus d'affinement un certain nombre de fois (3 par défaut).
- A l'aide des
![]() observés et des phases affinées, SnB calcule une densité
électronique par transformée de Fourier inverse, retournant
ainsi dans l'espace direct (figure 1).
observés et des phases affinées, SnB calcule une densité
électronique par transformée de Fourier inverse, retournant
ainsi dans l'espace direct (figure 1).
- Le programme extrait ensuite les plus grands pics
de la densité électronique, et ces pics sont considérés
comme des atomes. Ils remplacent le modèle initial, et seront utilisés
pour le calcul des facteurs de stucture lors du cycle suivant. Dans le
cas où la structure possède des atomes " lourds " (par exemples
des S) un nombre correspondant des plus grands pics extraits sera affecté
du facteur de diffusion atomique f adéquat.
| 3 - Utilisation pratique de SnB : |
|