Superposition of two molecules
The problem of superpositioning two (or more) molecules onto each other is a common task and one would assume it is also very simple. In fact, there exist several techniques to solve this problem, but there are a few details to consider which can make coding of the problem subtle. In the following I will discuss a FORTRAN program which superimposes two pdb files using the elegant quaternion method as described by S.K.Kearsley [1].
The superposition problem can be split up into two parts: A rotation around the geometric center of the molecule to bring it into the proper orientation, and a translation that superimposes the centers of the probe and target molecule(s).
The geometric center of the molecule can be found readily by averaging the x,y, and z coordinates of the n atoms k, respectively.
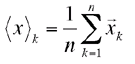
This center is not exactly the center of mass (we did not care what kind the atoms are, i.e., we did not calculate the moment). But a center defined this way will be the origin of the principal axes setting up the distance ellipsoid (similar to e.g., an anisotropic thermal parameter ellipsoid).
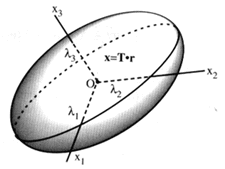
The (orthogonal) principal axes [2] can be obtained quite
easily by orthogonalization of the matrix A (i.e., the
matrix filled by the sum of the matrices of the metric tensor x
) :
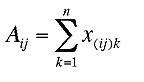 where
where 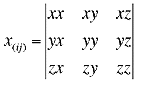
by a Jacobi transformation. The Jacobi transformation (or
orthogonalization) is an iterative application of rotations to a
matrix until all the off-diagonal values are zero at machine
precision [3]. If the matrix A
is symmetric (which A of course is) then a diagonal
matrix D exists so that
![]() where D has the eigenvalues in its diagonal :
where D has the eigenvalues in its diagonal : 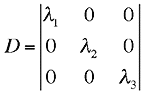
and P the normalized (orthogonal) eigenvectors of the principal axes in its columns. This orthogonalization is in fact equivalent to the solution of the eigenvalue problem [4]
![]() .
.
The feature of A is that it is real and symmetric
means that a solution with real eigenvectors and eigenvalues must
exist. This is good, but it is also the source of the mmm
symmetry of the ellipsoid, a nasty property as we'll see shortly.
Once we have determined the center of each molecule, we can shift
them to a common origin, usually (0,0,0). We are now faced with
the problem to find the rotation that gives the best
superposition of the molecules. What defines 'best'
superposition? One might be tempted to just rotate the principal
axes to an overlap. This could in principle work, and the
rotation matrix to overlap the axes can be easily calculated by a
Gauss-Jordan elimination [3] of
three 3x3 linear systems. This does give the correct answer in
cases where the molecules are pretty much pre-aligned, and the
molecules can also have a different number of atoms. But as a
result of the symmetry of the principal axes ellipsoid we have
lost information about the orientation of the axes: we need to
try 6 rotations (3 pairs of 180 deg rotated vectors) to find the
actual solution. To determine the best one, we are back to a
calculation of the pairwise distance r.m.s. (root mean square)
between atoms which we hoped to avoid in the first place.
So do we have to solve this as a least squares problem of r.m.s.
distances with all the problems associated (see, e.g. [1] for a discussion and further
reading)? No, not really. The above idea using the principal axes
rotation was quite elegant, but it needs to be improved. We need
to find a way to resolve the degeneracy of the principal axes
transformation. This can actually be done with an extension of
the 3x3 problem of the metric tensor matrix to a 4x4 matrix which
is constructed from a non-cyclic
permutation of the pairwise differences
between atom positions. The combination of elements follows the 4
constructors of the Quaternion group H, a small,
non-cyclic subgroup of GL2(C) [5].
We solve our linear quaternion eigenvalue system again
numerically by Jacobi transformation of the matrix Q
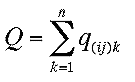 ,
where again
,
where again 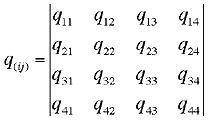
How the elements q are formed can be found in [1].
The smallest eigenvalue is the s.r.s.(sum of
residuals squared) for the best rotation and the associated
eigenvector contains element from which the 3x3 rotation matrix t
for the best superposition is constructed. The largest EV
contains the worst rotation, the associated eigenvalue the worst
s.r.s. The Quaternion method is a very elegant way to solve the
rotation problem and the r.m.s.d. least
squares minimization in one shot!
The application of the rotation and the back-translation of the
molecule are trivial final steps.
[1] S.K.Kearsley, On
the orthogonal transformation used for structural comparisons,
Acta Cryst. A45, 208 (1989)
[2] D.A.Danielson, Vectors and Tensors in Engineering
and Physics,
Addison-Wesley (1992), D.A.Sands, Vectors and Tensors in
Crystallography, Dover Publishing (1982)
[3] W.H.Press, S.A.Teukolsky, W.T.Vettering and B.P. Flannery, Numerical Recipes,
2nd edition, Cambridge University Press (1992)
[4] S. Lipschutz, Linear
Algebra, 2nd edition, Schaum Outline Series,
McGraw-Hill (1991)
[5] M.Artin, Algebra,
Prentice-Hall (1991)
[6] J.Hart, Quaternion
demonstrator (SGI only), Stanford University
![]() Back to X-ray Facility Introduction
Back to X-ray Facility Introduction
LLNL Disclaimer
This World Wide Web site conceived and maintained by
Bernhard Rupp (br@llnl.gov)
Last revised April 06, 1999 11:04
UCRL-MI-125269
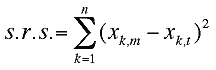{kind=link}
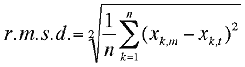{kind=link}