Superposition of two molecules
The problem of superpositioning two (or more) molecules onto each other is a common task and one would assume it is also very simple. In fact, there exist several techniques to solve this problem, but there are a few details to consider which can make coding of the problem subtle. In the following I will discuss a FORTRAN program which superimposes two pdb files using the elegant quaternion method.
The code that implements the quaternion method as described in [1] operates as follows :
First, two pdb files are opened and the pairwise correspondence of atoms is checked on several levels of increasing strictness : same number of atoms, same residues, same atom type. At any point a discrepancy is detected, one has the option to proceed or not, which may depend on the particulars of your problem.
In the next section, the program determines the centers of the molecules and creates the positve and negative pairwise differences of the atomic coordinates (all variables with *m refer to the moving probe, all with *f to the fixed target) are :
c --- sum up all coordinates (in dble precision) to find centre --- PDBS0131
do k=1,imov PDBS0132
do i=1,3 PDBS0133
sumf(i)=sumf(i)+xf(k,i) PDBS0134
summ(i)=summ(i)+xm(k,i) PDBS0135
end do PDBS0136
end do PDBS0137
do i=1,3 PDBS0138
cm(i)=sngl(summ(i)/imov) PDBS0139
cf(i)=sngl(sumf(i)/imov) PDBS0140
tr(i)=cf(i)-cm(i) PDBS0141
end do PDBS0142
PDBS0143
write(*,'(/a,3f8.3)')' Centre of target molecule =',(cf(i),i=1,3)PDBS0144
write(*,'(a,3f8.3)') ' Centre of moving molecule =',(cm(i),i=1,3)PDBS0145
write(*,'(a,3f8.3/)')' T - vector probe -> target =',(tr(i),i=1,3)PDBS0146
PDBS0147
write (*,'(a)')' Creating coordinate differences.......' PDBS0148
c --- create coordinate differences delta x plus (dxp) and minus (dxm) PDBS0149
do k=1,imov PDBS0150
do j=1,3 PDBS0151
dxm(k,j)=xm(k,j)-cm(j)-(xf(k,j)-cf(j)) PDBS0152
dxp(k,j)=xm(k,j)-cm(j)+(xf(k,j)-cf(j)) PDBS0153
end do PDBS0154
end do PDBS0155
Now we are ready to fill the quaternion matrix q as described by Kearsley with the pairwise coordinate differences we just obtained :
c --- fill upper triangle of (symmetric) quaternion matrix -- PDBS0157
write (*,'(a)')' Filling quaternion matrix ............' PDBS0158
do k=1,imov PDBS0159
c --- diags are sums of squared cyclic coordinate differences PDBS0160
q(1,1)=q(1,1)+dxm(k,1)**2+dxm(k,2)**2+dxm(k,3)**2 PDBS0161
q(2,2)=q(2,2)+dxp(k,2)**2+dxp(k,3)**2+dxm(k,1)**2 PDBS0162
q(3,3)=q(3,3)+dxp(k,1)**2+dxp(k,3)**2+dxm(k,2)**2 PDBS0163
q(4,4)=q(4,4)+dxp(k,1)**2+dxp(k,2)**2+dxm(k,3)**2 PDBS0164
c --- cross differences PDBS0165
q(1,2)=q(1,2)+dxp(k,2)*dxm(k,3)-dxm(k,2)*dxp(k,3) PDBS0166
q(1,3)=q(1,3)+dxm(k,1)*dxp(k,3)-dxp(k,1)*dxm(k,3) PDBS0167
q(1,4)=q(1,4)+dxp(k,1)*dxm(k,2)-dxm(k,1)*dxp(k,2) PDBS0168
q(2,3)=q(2,3)+dxm(k,1)*dxm(k,2)-dxp(k,1)*dxp(k,2) PDBS0169
q(2,4)=q(2,4)+dxm(k,1)*dxm(k,3)-dxp(k,1)*dxp(k,3) PDBS0170
q(3,4)=q(3,4)+dxm(k,2)*dxm(k,3)-dxp(k,2)*dxp(k,3) PDBS0171
end do PDBS0172
c --- fill the rest by transposing it onto itself PDBS0173
call trpmat(4,q,q) PDBS0174
write (*,'(/a)') PDBS0175
& ' q(1) q(2) q(3) q(4)' PDBS0176
do i=1,4 PDBS0177
write(*,'(4e13.5)') (q(i,j),j=1,4) PDBS0178
end do PDBS0179
The solution of the eigenvalue problem by Jacobi orthogonalization is straight forward using a canned routine from Numerical recipes :
c --- orthogonalization by jacobi rotation = solution of EV -problem -- PDBS0181
write (*,'(/a)')' Jacobi orthogonalization ..........' PDBS0182
n=4 PDBS0183
ns=4 PDBS0184
call jacobi(q,n,ns,dm,vm,nmrot) PDBS0185
c --- sort eigenvectors after eigenvalues, descending -- PDBS0186
write (*,'(a/)')' Sorting eigenvalues/vectors .......' PDBS0187
call eigsrt(dm,vm,n,ns) PDBS0188
write (*,'(a,i2,a)')' Eigenvalues and Eigenvectors (', PDBS0189
& nmrot,' Jacobi rotations)' PDBS0190
write (*,'(a)') ' e(1) e(2) e(4) e(4)' PDBS0191
write (*,'(4e12.5,i5)') (dm(j),j=1,4) PDBS0192
write (*,'(a)') ' ev(1) ev(2) ev(3) ev(4)' PDBS0193
do i=1,4 PDBS0194
write(*,'(4f12.6)') (vm(i,j),j=1,4) PDBS0195
end do PDBS0196
The smallest eigenvalue is the s.r.s.(sum of residuals squared) for the best rotation and the associated eigenvector contains element from which the 3x3 rotation matrix t for the best superposition is constructed.
c --- fill the rotation matrix which is made of elements from 4th EV PDBS0206
t(1,1)=vm(1,4)**2+vm(2,4)**2-vm(3,4)**2-vm(4,4)**2 PDBS0207
t(2,1)=2*(vm(2,4)*vm(3,4)+vm(1,4)*vm(4,4)) PDBS0208
t(3,1)=2*(vm(2,4)*vm(4,4)-vm(1,4)*vm(3,4)) PDBS0209
t(1,2)=2*(vm(2,4)*vm(3,4)-vm(1,4)*vm(4,4)) PDBS0210
t(2,2)=vm(1,4)**2+vm(3,4)**2-vm(2,4)**2-vm(4,4)**2 PDBS0211
t(3,2)=2*(vm(3,4)*vm(4,4)+vm(1,4)*vm(2,4)) PDBS0212
t(1,3)=2*(vm(2,4)*vm(4,4)+vm(1,4)*vm(3,4)) PDBS0213
t(2,3)=2*(vm(3,4)*vm(4,4)-vm(1,4)*vm(2,4)) PDBS0214
t(3,3)=vm(1,4)**2+vm(4,4)**2-vm(2,4)**2-vm(3,4)**2 PDBS0215
PDBS0216
do i=1,3 PDBS0217
write(*,'(3f11.5)') (t(i,j),j=1,3) PDBS0218
end do PDBS0219
The application of the rotation and the back-translation of the molecule are trivial final steps :
c --- reset dxm to store the individual rmsd's in it now - PDBS0221
call filmat(nat,3,dxm,0) PDBS0222
PDBS0223
c --- xm and xf are not translated - do now PDBS0224
do k=1,imov PDBS0225
c --- subtract cm PDBS0226
xm(k,1:3)=xm(k,1:3)-cm PDBS0227
c --- rotate it PDBS0228
call rotvec(3,xm(k,1:3),t) PDBS0229
c --- now add cf PDBS0230
xm(k,1:3)=xm(k,1:3)+cf PDBS0231
do i=1,3 PDBS0232
dxm(k,i)=sqrt((xf(k,i)-xm(k,i))**2) PDBS0233
end do PDBS0234
end do PDBS0235
PDBS0236
Finally, we create a new file and we can apply the same
transformation to another molecule, if desired.
![]() Get the program
(PDBSUP)
Get the program
(PDBSUP)
[1] S.K.Kearsley, On
the orthogonal transformation used for structural comparisons,
Acta Cryst. A45, 208 (1989)
[2] W.H.Press, S.A.Teukolsky, W.T.Vettering and B.P. Flannery, Numerical Recipes,
2nd edition, Cambridge University Press (1992)
![]() Back to X-ray Facility Introduction
Back to X-ray Facility Introduction
LLNL Disclaimer
This World Wide Web site conceived and maintained by
Bernhard Rupp (br@llnl.gov)
Last revised April 06, 1999 11:04
UCRL-MI-125269
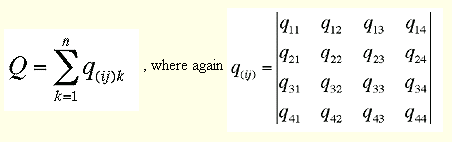{kind=link}
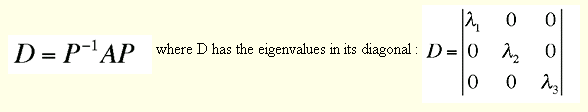{kind=link}
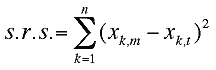{kind=link}