|
The Design of Structure Data | |
The design of the own structure files
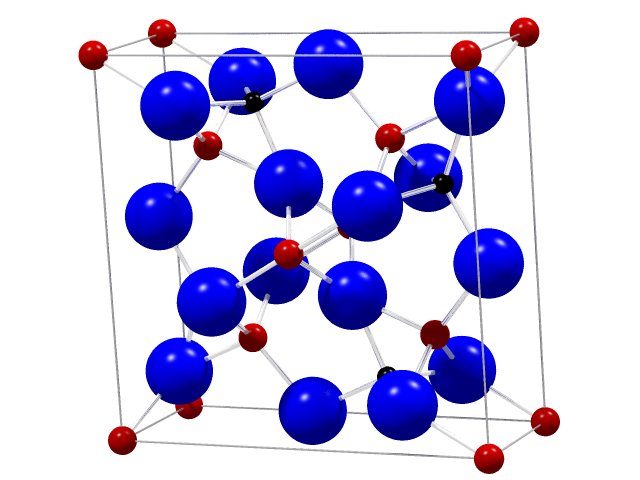
For the DOS version of PowderCell an individual structure data format was created in 1992 as we have started to develop this program. It contains besides the lattice constants the identifiers for the several positions, the atomic numbers, the atomic positions, the replacements and occupations, the isotropic Debye-Waller factors, the space-group number and the setting, if more than one exists. These data must be given for the asymmetric unit, exclusively. This form of structure data will be imported by the Windows version, too. Since someone likes it more to use a simple ASCII editor for the creation of structure files in the following table an example will be given:
|
The first line starts with the keyword CELL. The following six numbers define the lattice constants ao, bo, co in Å and a, b und g in degrees. These numbers always must be entered, even then, if two or three values are identically (e.g. in the cubic system ao = bo = co and a = b = g = 90°). In the given example the structure will be described within the orthorhombic crystal system. Therefore, all angles are 90° whereas the length of the basis vectors are different. All other lines except the last one with the keyword RGNR define the atomic positions of the content of the asymmetric unit. Usually, these data can be found in structure reports or will be given in several data bases. Whereas in principle the data can be entered unformatted, for the identifier a well-defined limitation exists. So the program interprets the first four characters of each line as identifier of this position. That means that the respective atomic number which follows the identifier must be given later. The atomic number defines all these atom-specific properties like relative mass, atomic scattering factor, mass attenuation, anomaleous dispersion etc. The next three numbers define the relative atomic position (x,y,z). The coordinates can be given also outside the unit-cell dimension i.e. the coefficients may be higher than 1, absolutely. The identifier, the atomic number and the coordinate are necessary data for each position. However, one may add further information like occupation, replacement (substitution) and an isotropic Debye-Waller factor. Of course, if only the Debye-Waller factors shall be entered also the so-called multiplied substitution and replacement factor (SOF) must be inserted as 1. Otherwise it is not necessary to enter these data for all positions.
If a replacement shall be considered wether an identifier nor the coordinates must be given. The program recognizes the blanks in the beginning of the line and interprets the first value as atomic number, the second as SOF and the possible third as Debye-Waller factor. The coordinates will be taken from the position before. Please notice that the program doesn't check the size of SOF automatically. Usually, the sum of all SOF's of a single position should be equal or smaller than 1. The sum corresponds with the occupation of this position. 1 is equivalent to 100%.
Since the program uses the asymmetric unit it needs information about the structure symmetry. This will be arranged by the line which starts with the keyword RGNR. This is the abbreviation of the german word "Raumgruppen-Nummer" which means space-group number. Unfortunately, sometimes there exists more than one setting of a space-group type. Thus, a further number must be given if the structure hasn't been described using a conventional setting (standard setting). For more information please use the detailed help of the program or the listing of used settings.
At least an arbitrary comment can be entered. However, in version 1.0 it will be lost if the structure file will be imported and saved again. In version 2.0 a special comment editor has been implemented.
Generally, the files must have the extension *.cel.
Additionally to the very compact but individual *.cel-files, PowderCell is able to import crystal structure data in form of exported ICSD files as well as SHELX files. In contrast to the own crystal structure data files the use of ICSD files enables the consideration of anisotropic temperature factors. Thus in version 2.0 a conversion of anisotropic to equivalent Debye-Waller factors has been implemented.
Please notice that the program enables not only the use of conventional settings described detailed in the International Tables for Crystallography, Vol.A, but also of a lot of different unit cell definitions. However, in a small number of structure determinations sometimes an additional origin shift has been used, i.e. all atomic positions of the asymmetric unit contain this shift, too. This hasn't been considered within PowderCell 1.0 and the shift must be substracted manually. In version 2.0 an additional translation can be defined which supports an automatical shift of all positions of the asymmetric unit.
If the data conversion cannot finished successfully because of a unknown structure setting the program takes all positions but defines the structure symmetry as P 1.
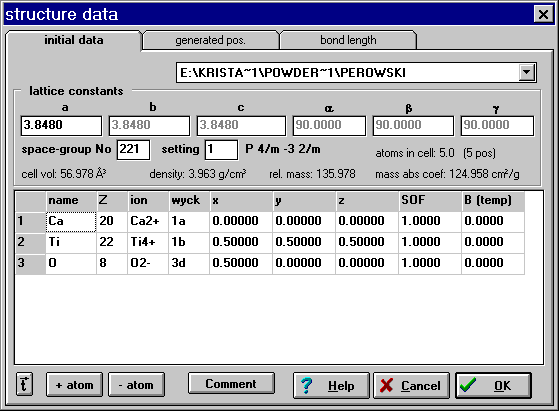 In PowderCell it is possible to enter a complete or to edit a previously imported structure file.
In PowderCell it is possible to enter a complete or to edit a previously imported structure file.
The inserted editor makes it more easy to check the entered data, e.g. it considers the used space-group symmetry. If a cubic space-group number has been entered only one lattice constant must be defined. All other will be copied by the program itself. This will be shown by the gray-colored numbers. However, please notice that this requires the input of the space-group number at first. On the other hand the input of the atomic coordinates is more easy than using an ASCII editor. So only the Wyckoff letter must be inserted and if the position hasn't a degree of freedom all coefficient (x,y and z) will be inserted by the program immediately. Of course, undefined coefficients must be entered manually. In contrast to POWDER CELL's structure files (*.cel) a manually created file contains additional information. Whereas the DOS version of PowderCell only defines atoms in the structure data file in form of the atomic number, the Windows version considers also several ionic states of atoms. So the use of different bonding states of one and the same element (e.g. Fe2+ and Fe3+ in Fe3O4) can be considered simultaneously in the actual version.
Furthermore the screen contains some derived properties, e.g. the unit-cell volume, the relative mass of the cell content and the number of generated as well as really occupied positions. This supports to detect input errors. Since this data will be calculated it don't will be actualized immediately. That applies to the atomic position, too. Only if the window is closed by the OK button all old data really will be overwritten by the actualized values and the structure will be generated again. The calculated values can be seen if the screen will be opened again.
Federal Institute for Materials Research and Testing
Unter den Eichen 87, D-12205 Berlin,
Germany